Plot
Heatmap
A heatmap can be used to show relative abundances oftaxa in samples or groups of samples. The plot.heatmap function can be used like this.
[1]:
import qdiv
obj = qdiv.MicrobiomeData.load_example("Saheb-Alam_DADA2") #First we load example data
obj.rename_features(inplace=True, name_type="ASV") #This is the name the features ASV1, ASV2
obj.tax_prefix(add=True, inplace=True) #This is to add prefix to the taxonomic classified, i.e., d__ for domain, p__ for phylum, etc.
print(obj.meta) #Let's have a look at the meta data before plotting the heatmap
location feed mfc
sample
S4 anode acetate B
S5 anode acetate B
S6 anode acetate B
S7 anode acetate B
S10 cathode acetate B
S11 cathode acetate B
S12 cathode acetate B
S13 cathode acetate B
S20 anode glucose D
S21 anode glucose D
S22 anode glucose D
S23 anode glucose D
S26 cathode glucose D
S27 cathode glucose D
S28 cathode glucose D
S29 cathode glucose D
[2]:
fig, ax, data = qdiv.plot.heatmap(obj, group_by=["feed", "location"], levels=["Phylum", "Genus"])
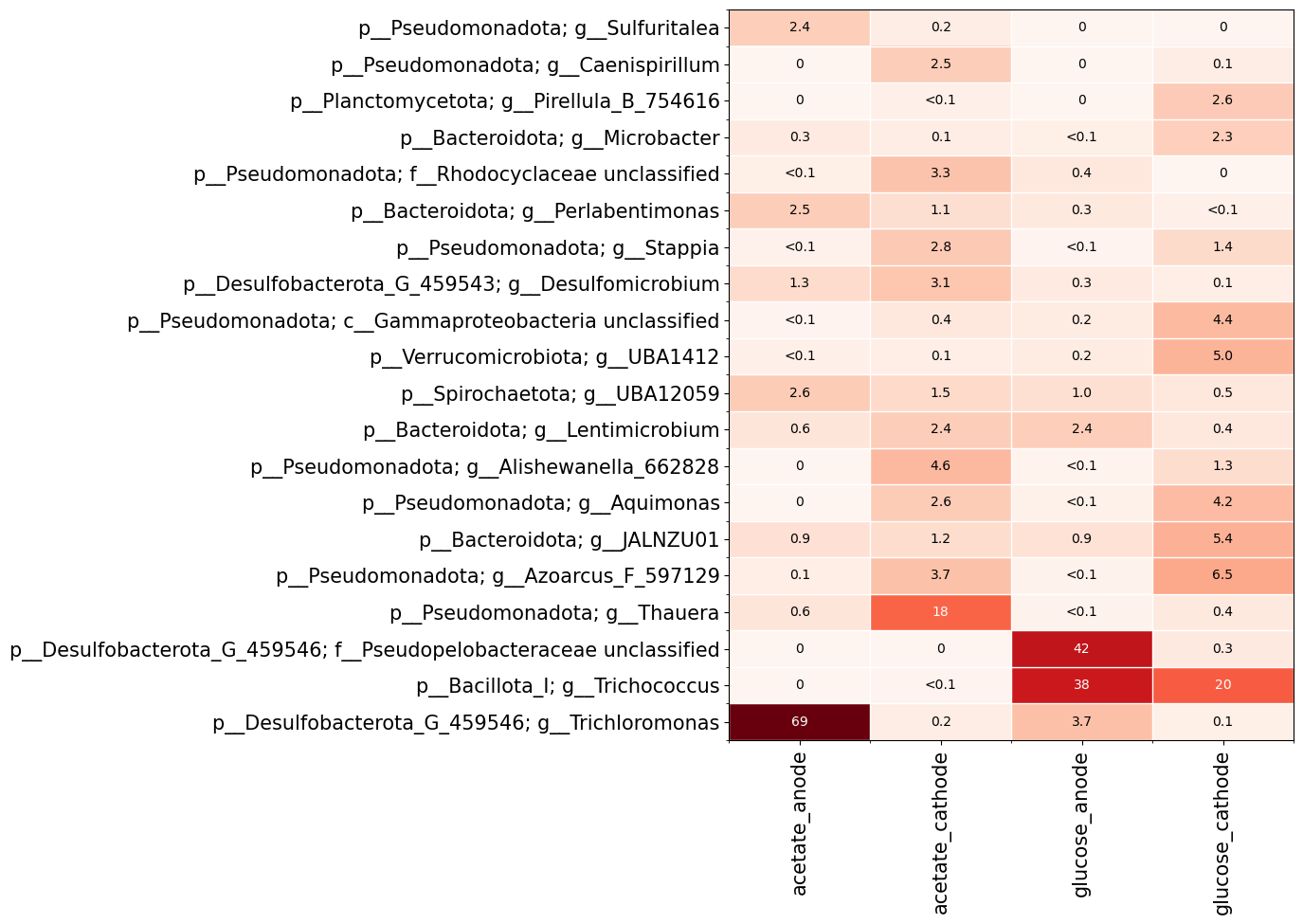
Here I chose to group the samples based on the meta data columns ‘feed’ and ‘location’. I also specified that I want to show phylum and genus levels on the y-axis. The features are grouped based on the lowest taxonomic level chosen (i.e. genus in this case).
Alpha diversity profiles
The plot.alpha_diversity_profile function let’s us visualize how alpha diversity depends on diversity order.
[3]:
import qdiv
obj = qdiv.MicrobiomeData.load_example("Saheb-Alam_DADA2")
obj.rename_features(inplace=True, name_type="ASV") #This is the name the features ASV1, ASV2
obj.tax_prefix(add=True, inplace=True) #This is to add prefix to the taxonomic classified, i.e., d__ for domain, p__ for phylum, etc.
obj.rarefy(inplace=True) #Rarefy to make comparison of alpha diversity between samples easier
fig, ax, data = qdiv.plot.alpha_diversity_profile(obj, color_by="location")
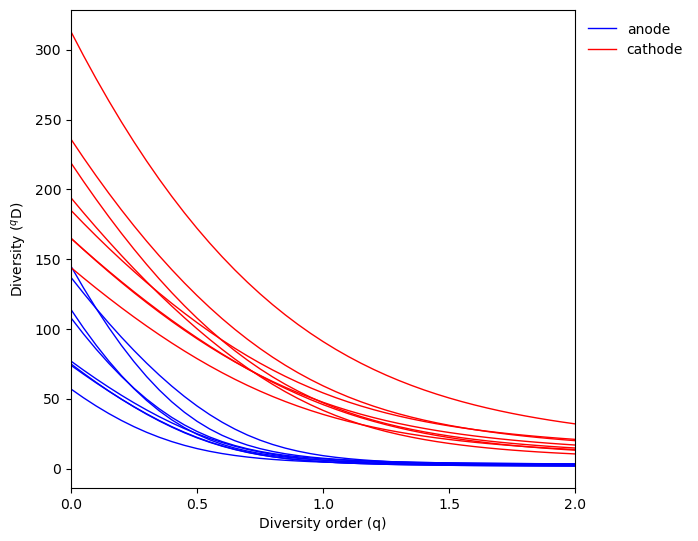
Here we can see the cathode samples tend to have higher diversity than anode samples for all diversity orders.
Beta diversity
Similarities and differences in community composition between samples if often visualized using an ordination. First, we calculate pairwise dissimilarities between samples using the diversity.naive_beta function.
[4]:
import qdiv
obj = qdiv.MicrobiomeData.load_example("Saheb-Alam_DADA2")
obj.rename_features(inplace=True, name_type="ASV") #This is the name the features ASV1, ASV2
obj.tax_prefix(add=True, inplace=True) #This is to add prefix to the taxonomic classified, i.e., d__ for domain, p__ for phylum, etc.
obj.rarefy(inplace=True)
dis = qdiv.diversity.naive_beta(obj, q=1) #Here I calculate for q=1
Next, we plot a principal coordinate analysis using the plot.ordination function.
[5]:
fig, ax, res = qdiv.plot.ordination(dis, obj, color_by="location", shape_by="feed")
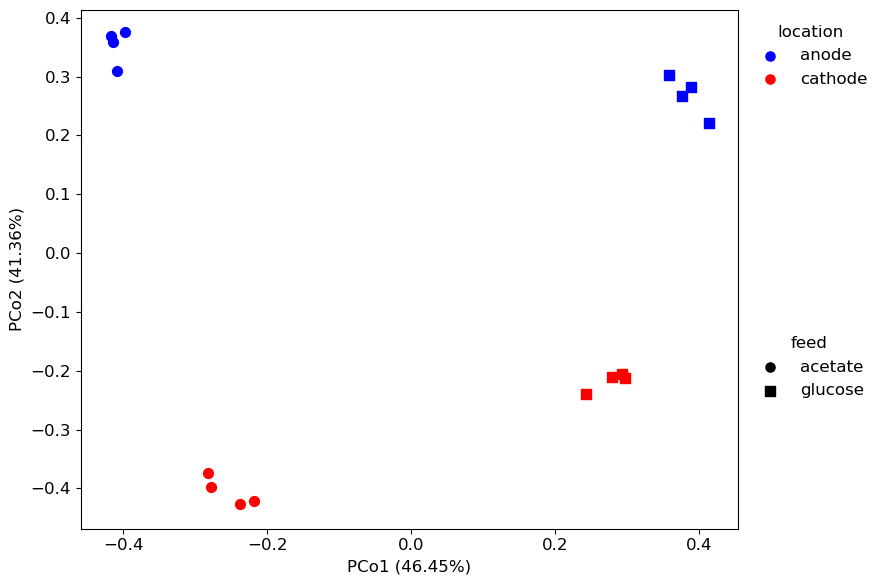
The res output is a dictionary that includes information about the ordination results.
res[“meta”] contains the metadata with the coordinates for the points added as columns: “PCo1” and “PCo2”.
res[“pct_explained”] contains information about the percent variation explained by each axis.
[7]:
print(res["meta"])
print(res["pct_explained"])
location feed mfc PCo1 PCo2
S4 anode acetate B -0.422924 -0.366974
S5 anode acetate B -0.414454 -0.308858
S6 anode acetate B -0.419395 -0.356684
S7 anode acetate B -0.404560 -0.370697
S10 cathode acetate B -0.231987 0.431153
S11 cathode acetate B -0.270401 0.400800
S12 cathode acetate B -0.212815 0.419261
S13 cathode acetate B -0.272921 0.380127
S20 anode glucose D 0.414208 -0.223915
S21 anode glucose D 0.381922 -0.262631
S22 anode glucose D 0.391101 -0.284268
S23 anode glucose D 0.358608 -0.309530
S26 cathode glucose D 0.277438 0.207323
S27 cathode glucose D 0.295947 0.208371
S28 cathode glucose D 0.294605 0.199813
S29 cathode glucose D 0.235628 0.236709
PCo1 46.34
PCo2 41.23
PCo3 10.28
PCo4 1.32
PCo5 0.42
PCo6 0.22
PCo7 0.09
PCo8 0.08
PCo9 0.03
dtype: float64
[ ]: